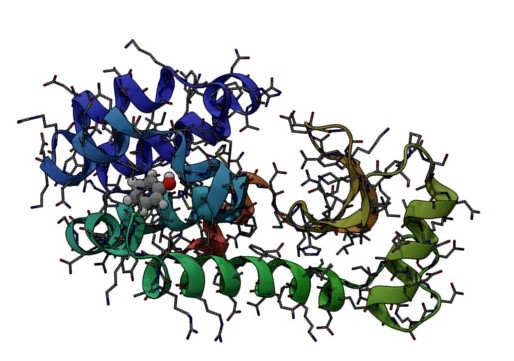
This investigation is motivated by the need to investigate if first-principles quantum mechanical (QM) calculations on entire protein-ligand systems are now computationally tractable and a promising avenue of future research. QM-based binding free energy calculations are not sufficiently explored due to a community perception of intractable computational cost and difficult convergence. We demonstrate that with moderate access to modern HPC resources, protein-ligand free energies of binding can be calculated at the quantum-mechanical level using linear-scaling DFT. In our 2600 atom protein-ligand system, full-DFT calculations on snapshots converge at the same rate as in the traditional force field based MM-PBSA approach. We test three exchange-correlation functionals and different empirical dispersion corrections and take an in depth look at the convergence and errors in both the enthalpy and entropy. With our benchmarks we can give clear recommendations how QM-PBSA should be performed for comparable systems. This opens up new avenues for impact in areas of current technological and societal interest such as drug optimisation and biomolecular association where the required chemical accuracy often requires one to go beyond classical mechanical and/or empirical approaches in order to achieve predictive conclusions. Such calculations with ONETEP can also be used to train Machine-Learned (ML) force fields bespoke for particular proteins to enable long reactive MD simulations with DFT accuracy.
Protein-Ligand Free Energies of Binding from Full-Protein DFT Calculations